ท้าทายการครอบงำของ Google DeepMind ในด้านชีววิทยาการคำนวณ นักวิจัยจาก Harvard Medical School ได้เปิดตัว popEVE ซึ่งเป็นโมเดลปัญญาประดิษฐ์ใหม่ที่ออกแบบมาเพื่อวินิจฉัยโรคทางพันธุกรรมที่หายากและมีความจำเพาะที่เพิ่มขึ้น
เผยแพร่ในวันนี้ใน Nature Genetics เครื่องมือนี้ได้รวมข้อมูลประชากรมนุษย์เพื่อลดการคาดการณ์เชิงบวกที่ผิดพลาดอย่างมาก ซึ่งเป็นข้อบกพร่องที่ยังคงมีอยู่ในโมเดลที่มีอยู่ เช่น AlphaMissense
โดยการปรับเทียบความรุนแรงของตัวแปรทั่วทั้งระบบ proteome นั้น popEVE ประสบความสำเร็จในการระบุยีนที่มีศักยภาพ 123 ยีนสำหรับความผิดปกติของพัฒนาการ ซึ่งถือเป็นความก้าวหน้าในการวินิจฉัยสำหรับผู้ป่วยที่ยังไม่ได้รับการแก้ไขแม้ว่าจะมีการทดสอบอย่างกว้างขวางก็ตาม
popEVE ตั้งเป้าที่จะแก้ไขปัญหาผลบวกลวง
แม้จะมีการขยายตัวอย่างรวดเร็วของจีโนม การจัดลำดับในการตั้งค่าทางคลินิก ผลการวินิจฉัยสำหรับความผิดปกติทางพันธุกรรมที่หายากยังคงต่ำอย่างดื้อรั้น โดยบางกลุ่มพบว่าเพียง 25% ของ probands ได้รับการวินิจฉัยทางพันธุกรรมขั้นสุดท้าย
แพทย์มักเผชิญกับ”ตัวแปรที่มีนัยสำคัญไม่แน่นอน”(VUS) มากมายหลากหลาย ซึ่งการเปลี่ยนแปลงทางพันธุกรรมที่มีผลกระทบต่อสุขภาพของมนุษย์ไม่เป็นที่ทราบ
ความคลุมเครือนี้ทำให้เกิดปัญหาคอขวดในการวินิจฉัย โดยการระบุตัวแปรเฉพาะที่รับผิดชอบต่อ อาการของผู้ป่วยต้องใช้เวลานานและมักไร้ผล การตีความในปัจจุบันมักจะล้มเหลวในการแยกแยะความแตกต่างระหว่างตัวแปรที่ทำให้เกิดความผิดปกติที่รุนแรงในเด็กและที่มีผลกระทบเล็กน้อยซึ่งจะปรากฏในภายหลังในชีวิตเท่านั้น ซึ่งเป็นความแตกต่างที่สำคัญสำหรับการดูแลเด็ก
ตามรายงานการวิจัย popEVE จัดการกับช่องว่างที่แม่นยำนี้ด้วยการบังคับใช้เกณฑ์ที่เข้มงวดมากขึ้นสำหรับ การเกิดโรค ในการทดสอบ แบบจำลองนี้แสดงให้เห็นถึงการลดลงอย่างมากในการทำนายผลบวกลวงภายในประชากรทั่วไป โดยระบุว่ามีเพียง 11% ของบุคคลที่เป็นพาหะของตัวแปรที่รุนแรง
ระดับความจำเพาะนี้เป็นการปรับปรุงที่โดดเด่นเหนือเครื่องมือล้ำสมัยที่มีอยู่ ตัวอย่างเช่น AlphaMissense ของ Google DeepMind จัดหมวดหมู่ประมาณ 44% ของประชากรทั่วไปว่ามีตัวแปรที่รุนแรงใกล้เคียงกันโดยมีเกณฑ์การเรียกคืนที่เทียบเคียงได้ ด้วยการกรองสัญญาณรบกวนนี้ออก popEVE ช่วยให้แพทย์มุ่งเน้นไปที่ตัวแปรที่มีแนวโน้มว่าจะเป็นสาเหตุมากที่สุด
ประสิทธิภาพของแบบจำลองได้รับการตรวจสอบอย่างเข้มงวดในกลุ่มผู้ป่วย 31,058 รายที่มีความผิดปกติด้านพัฒนาการขั้นรุนแรง (SDD) ซึ่งได้มาจากการศึกษา Deciphering Developmental Disorders (DDD), GeneDx และ Radboud University Medical Center
ภายในขอบเขตที่ครอบคลุมนี้ ชุดข้อมูล ซึ่งเป็นเกณฑ์ความรุนแรงที่มีความมั่นใจสูงของ popEVE (ตั้งไว้ที่-5.056) เผยให้เห็นการเพิ่มประสิทธิภาพของตัวแปรที่ทำให้เกิดโรคถึง 15 เท่า ซึ่งสูงกว่าวิธีการชั้นนำอื่นๆ เช่น PrimateAI-3D ถึงห้าเท่า พลังทางสถิตินี้ช่วยให้แบบจำลองสามารถให้การวินิจฉัยได้สำเร็จสำหรับประมาณหนึ่งในสามของกรณีที่ก่อนหน้านี้ฝ่าฝืนคำอธิบายภายใต้โปรโตคอลการทดสอบมาตรฐาน
บางทีสิ่งที่สำคัญที่สุดสำหรับสาขาพันธุศาสตร์ทางการแพทย์ก็คือความสามารถของแบบจำลองในการเปิดเผยความสัมพันธ์ของโรคใหม่ทั้งหมด การวิเคราะห์ระบุยีนผู้สมัครใหม่ 123 ยีนที่เชื่อมโยงกับความผิดปกติของพัฒนาการ โดย 119 ยีนสามารถระบุได้ในระดับตัวแปรเดียว
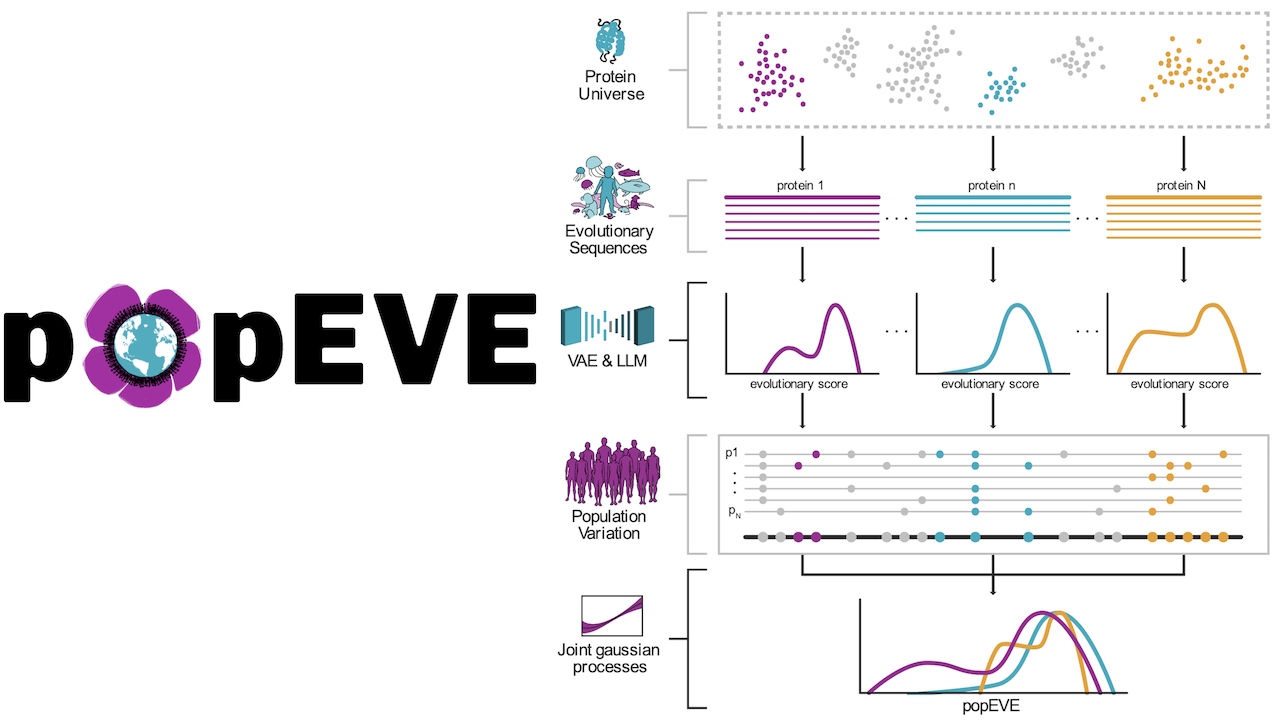
แบบจำลองทั้งโปรตีโอมสำหรับพันธุกรรมของโรคในมนุษย์
(ที่มา: ธรรมชาติ – CC BY-NC-ND 4.0)
โดยเฉพาะอย่างยิ่ง ยีน 31 ตัวในจำนวนนี้ได้รับการกู้คืนโดยใช้ตัวแปรแบบ missense เพียงอย่างเดียว ซึ่งเป็นหมวดหมู่ของการกลายพันธุ์ที่โดยทั่วไปต้องใช้ข้อมูลที่ยืนยันการสูญเสียฟังก์ชัน (LoF) จึงจะได้รับการพิจารณาวินิจฉัย ความสามารถนี้ชี้ให้เห็นว่า popEVE สามารถตรวจจับสัญญาณที่ทำให้เกิดโรคซึ่งวิธีการเสริมคุณค่าแบบดั้งเดิมพลาดไป
การตรวจสอบความถูกต้องของการค้นพบเหล่านี้ให้ผลลัพธ์ทางคลินิกแล้ว นับตั้งแต่เริ่มการศึกษา ยีนใหม่จำนวน 25 ยีนจากทั้งหมด 123 ยีนได้รับการยืนยันอย่างเป็นอิสระจากห้องปฏิบัติการอื่นๆ และได้เพิ่มอย่างเป็นทางการในฐานข้อมูล Developmental Disorder Gene to Phenotype (DDG2P) แล้ว
นอกจากนี้ เมื่อนำไปใช้กับการกลายพันธุ์แบบ de novo missense (DNM) แบบจำลองจะทำเครื่องหมาย 7% ของตัวแปรในกรณีที่มีความรุนแรง เทียบกับเพียง 0.5% ในกลุ่มควบคุมที่ดี ซึ่งแสดงให้เห็นถึงการแยกในระดับสูงระหว่าง รูปแบบที่ก่อให้เกิดโรคและไม่เป็นพิษเป็นภัย
Debora Marks ศาสตราจารย์ด้านชีววิทยาระบบที่ Harvard Medical School เน้นย้ำว่าเครื่องมือนี้ได้รับการออกแบบมาเพื่อแปลผลที่ได้รับทางสถิติเหล่านี้ให้เป็นผลลัพธ์ทางคลินิกที่จับต้องได้ “เป้าหมายของเราคือการพัฒนาแบบจำลองที่จัดอันดับตัวแปรต่างๆ ตามความรุนแรงของโรค โดยให้มุมมองจีโนมของบุคคลที่มีการจัดลำดับความสำคัญและมีความหมายทางคลินิก”
การปรับเทียบโปรตีโอม
แบบจำลองล้ำสมัยก่อนหน้านี้ ซึ่งรวมถึง EVE และ AlphaMissense นั้นมีความเป็นเลิศในการจัดอันดับตัวแปรต่างๆ ภายในยีนเดียว แต่ประสบปัญหาในการเปรียบเทียบความรุนแรงของยีนต่างๆ ด้วยเหตุนี้ คะแนนที่สูงมักปรากฏสำหรับตัวแปรที่รบกวนการทำงานของโปรตีน แต่ไม่จำเป็นต้องทำให้เกิดโรคร้ายแรงในบริบทของมนุษย์
popEVE แก้ปัญหานี้โดยการรวมข้อมูลวิวัฒนาการเชิงลึก (โดยใช้ EVE และแบบจำลองภาษา ESM-1v) เข้ากับข้อจำกัดของประชากรมนุษย์ เพื่อระบุตัวแปรที่ยอมรับได้ตามธรรมชาติ ทีมงานใช้ข้อมูลจาก UK Biobank (UKBB) และ gnomAD v2
มีการใช้กระบวนการเกาส์เซียนแฝงเพื่อปรับเทียบคะแนนวิวัฒนาการเทียบกับการเปลี่ยนแปลงของมนุษย์ที่สังเกตได้นี้ ทำให้เกิดคะแนน”ความลบล้าง”ที่เป็นหนึ่งเดียว ด้วยการปรับเปลี่ยนนี้ ความก้าวหน้าทางคลินิกครั้งสำคัญจึงเกิดขึ้นได้: การวิเคราะห์แบบ”ซิงเกิลตัน”ซึ่งสามารถจัดลำดับความสำคัญของตัวแปรเชิงสาเหตุได้โดยใช้เพียงตัวอย่างของเด็กเท่านั้น
วิธีการแบบดั้งเดิมโดยทั่วไปต้องใช้การจัดลำดับแบบ”สาม”(พ่อแม่ + เด็ก) เพื่อระบุการกลายพันธุ์เดอโนโว ซึ่งเป็นกระบวนการที่มักมีราคาแพงมากหรือเป็นไปไม่ได้ในเชิงลอจิสติกส์
Mafalda Dias นักวิจัยที่ Center for Genomic Regulation, เน้นถึงผลกระทบในทางปฏิบัติของความสามารถนี้ “คลินิกไม่สามารถเข้าถึง DNA ของผู้ปกครองได้เสมอไปและผู้ป่วยจำนวนมากมาคนเดียว popEVE สามารถช่วยแพทย์เหล่านี้ระบุการกลายพันธุ์ที่ก่อให้เกิดโรคได้”
AlphaMissense ที่ท้าทาย
AlphaMissense ของ Google DeepMind ซึ่งเปิดตัวในเดือนกันยายน 2023 ก่อนหน้านี้ได้กำหนดมาตรฐานใหม่ด้วยการจัดหมวดหมู่ 89% ของรูปแบบ missense ที่เป็นไปได้ทั้งหมด อย่างไรก็ตาม ทีมงานของ Harvard ให้เหตุผลว่าแม้ว่า AlphaMissense จะมีความแม่นยำในด้านความคงตัวของโปรตีน แต่ก็ขาดการสอบเทียบทางคลินิกที่จำเป็นสำหรับการวินิจฉัย
การวิเคราะห์ทางสถิติแสดงให้เห็นว่า AlphaMissense คาดการณ์ตัวแปร”ที่ทำให้เกิดโรค”โดยเฉลี่ยห้าตัวแปรต่อคนโดยเฉลี่ย ในขณะที่ popEVE คาดการณ์น้อยกว่าหนึ่งตัว ความคลาดเคลื่อนดังกล่าวมีความสำคัญต่อการตั้งค่าทางคลินิก โดยที่การคาดการณ์มากเกินไปอาจนำไปสู่การวินิจฉัยผิดพลาดและความวิตกกังวลที่ไม่จำเป็น
รายงานของ PrpopEVE หมายเหตุเพิ่มเติม:
“popEVE ระบุยีน 442 ยีนในกลุ่มที่มีความผิดปกติด้านพัฒนาการ ซึ่งรวมถึงหลักฐานของผู้สมัครใหม่ 123 ราย ซึ่งหลายรายไม่จำเป็นต้องเสริมคุณค่าทั่วทั้งกลุ่ม”
“สุดท้ายนี้ เราแสดงให้เห็นว่าการค้นพบเหล่านี้ สามารถทำซ้ำได้จากการวิเคราะห์ผู้ป่วยภายนอกเพียงอย่างเดียว ซึ่งแสดงให้เห็นว่า popEVE มอบช่องทางใหม่สำหรับการวิเคราะห์ทางพันธุกรรมในสถานการณ์ที่วิธีการแบบเดิมล้มเหลว”
แม้ว่าประสิทธิภาพจะเพิ่มขึ้น แต่ popEVE ยังคงเป็นเครื่องมือในการวิจัยและยังไม่ได้รับการอนุมัติจาก FDA เพื่อใช้เป็นเครื่องมือวินิจฉัยแบบสแตนด์อโลน Marks Lab กำลังทำให้โมเดลพร้อมใช้งานผ่านพอร์ทัล popEVE แบบเปิดและพื้นที่เก็บข้อมูล popEVE ซึ่งตรงกันข้ามกับลักษณะที่เป็นกรรมสิทธิ์ของเครื่องมือด้านสุขภาพ AI เชิงพาณิชย์
การใช้งานในอนาคตขยายไปไกลกว่าการวินิจฉัยไปจนถึงการค้นพบยา เนื่องจากโมเดลสามารถระบุสาเหตุของโรคที่เฉพาะเจาะจงได้ กลไกภายในโครงสร้างโปรตีน
Rose Orenbuch นักวิจัยใน Marks Lab แสดงการมองโลกในแง่ดีเกี่ยวกับการบูรณาการของเครื่องมือเข้ากับขั้นตอนการทำงานทางคลินิก “ฉันรู้สึกว่าเราเข้าใกล้ PopEVE มากขึ้นอีกก้าวหนึ่งในการเป็นประโยชน์ในการพยายามวินิจฉัยโรคทางพันธุกรรมได้เร็วขึ้น”