Kwestionując dominację Google DeepMind w biologii obliczeniowej, naukowcy z Harvard Medical School zaprezentowali popEVE, nowy model sztucznej inteligencji przeznaczony do diagnozowania rzadkich chorób genetycznych ze zwiększoną swoistością.
Opublikowane dziś w Nature Genetics narzędzie integruje dane dotyczące populacji ludzkiej, aby radykalnie ograniczyć fałszywe przewidywania pozytywne, co stanowi stałą wadę istniejących modeli, takich jak AlphaMissense.
Poprzez kalibrację ciężkości wariantów w całym proteomie, popEVE z powodzeniem zidentyfikowało 123 nowe geny kandydujące na zaburzenia rozwojowe, zapewniając przełom w diagnostyce dla pacjentów, których wynik pozostał nierozwiązany pomimo szeroko zakrojonych badań.
popEVE ma na celu rozwiązanie problemu fałszywie dodatniego
Pomimo szybkiego rozwoju po sekwencjonowaniu genomu w warunkach klinicznych, skuteczność diagnostyki rzadkich zaburzeń genetycznych pozostaje stale niska, a w niektórych kohortach tylko 25% probantów otrzymuje ostateczną diagnozę genetyczną.
Klinicyści często mają do czynienia z szeroką gamą „wariantów o niepewnym znaczeniu” (VUS), czyli zmian genetycznych, których wpływ na zdrowie człowieka jest nieznany.
Ta niejednoznaczność tworzy wąskie gardło diagnostyczne, polegające na identyfikacji konkretnego wariantu odpowiedzialnego za stan pacjenta staje się przedsięwzięciem czasochłonnym i często bezowocnym. Obecna interpretacja często nie rozróżnia wariantów powodujących poważne zaburzenia rozpoczynające się w dzieciństwie od wariantów o umiarkowanych skutkach, które ujawniają się dopiero w późniejszym życiu, co jest zasadniczą różnicą w opiece pediatrycznej.
Zgodnie z dokument badawczy, popEVE wypełnia tę lukę w precyzji, wymuszając bardziej rygorystyczny próg dla patogeniczność. W testach model wykazał radykalne zmniejszenie liczby fałszywie pozytywnych przewidywań w populacji ogólnej, wskazując zaledwie 11% osób jako nosicieli ciężkich wariantów.
Ten poziom szczegółowości stanowi wyraźną poprawę w porównaniu z istniejącymi, najnowocześniejszymi narzędziami; na przykład badanie AlphaMissense opracowane przez Google DeepMind klasyfikuje około 44% ogólnej populacji jako nosicieli podobnie poważnych wariantów przy porównywalnym progu przypominania. Odfiltrowując te szumy, popEVE umożliwia klinicystom skupienie się na wariantach, które najprawdopodobniej mają związek przyczynowy.
Skuteczność modelu została rygorystycznie zweryfikowana na metakohorcie 31 058 pacjentów z ciężkimi zaburzeniami rozwojowymi (SDD), pochodzącej z badania Deciphering Developmental Disorders (DDD), GeneDx i Centrum Medycznego Uniwersytetu Radboud.
W ramach tego obszernego zbioru danych: Wysoce ufny próg istotności popEVE (ustawiony na-5,056) ujawnił 15-krotne wzbogacenie wariantów patogennych – pięć razy więcej niż w przypadku innych wiodących metod, takich jak PrimateAI-3D. Ta moc statystyczna umożliwiła modelowi skuteczne postawienie diagnozy w przypadku około jednej trzeciej przypadków, które wcześniej nie dawały się wyjaśnić w ramach standardowych protokołów testowych.
Być może najbardziej znacząca w dziedzinie genetyki medycznej jest zdolność modelu do odkrywania zupełnie nowych powiązań chorobowych. W analizie zidentyfikowano 123 nowe geny kandydujące powiązane z zaburzeniami rozwojowymi, z których 119 można było zidentyfikować na poziomie pojedynczego wariantu.
Ogólnoproteomowy model genetyki chorób człowieka
(Źródło: Nature – CC BY-NC-ND 4.0)
Warto zauważyć, że 31 z tych genów odzyskano przy użyciu samych wariantów zmiany sensu – kategorii mutacji, która zazwyczaj wymaga potwierdzenia danych dotyczących utraty funkcji (LoF), aby można ją było uznać za diagnostyczną. Zdolność ta sugeruje, że popEVE może wykryć sygnały chorobotwórcze, których nie dostrzegają tradycyjne metody oparte na wzbogacaniu.
Walidacja tych odkryć już przynosi wyniki kliniczne. Od rozpoczęcia badania 25 ze 123 nowych genów kandydujących zostało niezależnie potwierdzonych przez inne laboratoria i formalnie dodano do bazy danych Developmental Disorder Gene to Phenotype (DDG2P).
Co więcej, po zastosowaniu do mutacji zmiany sensu (DNM) de novo model wskazał 7% wariantów w przypadkach tak poważnych, w porównaniu z zaledwie 0,5% w zdrowej grupie kontrolnej, co wskazuje na wysoki stopień separacji między patogennymi i zmiany łagodne.
Debora Marks, profesor biologii systemowej w Harvard Medical School, podkreśliła, że narzędzie to ma na celu przełożenie tych korzyści statystycznych na wymierne wyniki kliniczne. „Naszym celem było opracowanie modelu, który szereguje warianty według ciężkości choroby – zapewniając priorytetowy, klinicznie znaczący obraz genomu danej osoby.”
Kalibracja proteomu
Poprzednie najnowocześniejsze modele, w tym EVE i AlphaMissense, doskonale radzą sobie z rankingiem wariantów w obrębie jednego genu, ale mają trudności z porównaniem ciężkości różnych genów. W związku z tym często wysokie wyniki uzyskują warianty, które zakłócają funkcję białek, ale niekoniecznie powodują poważną chorobę u człowieka.
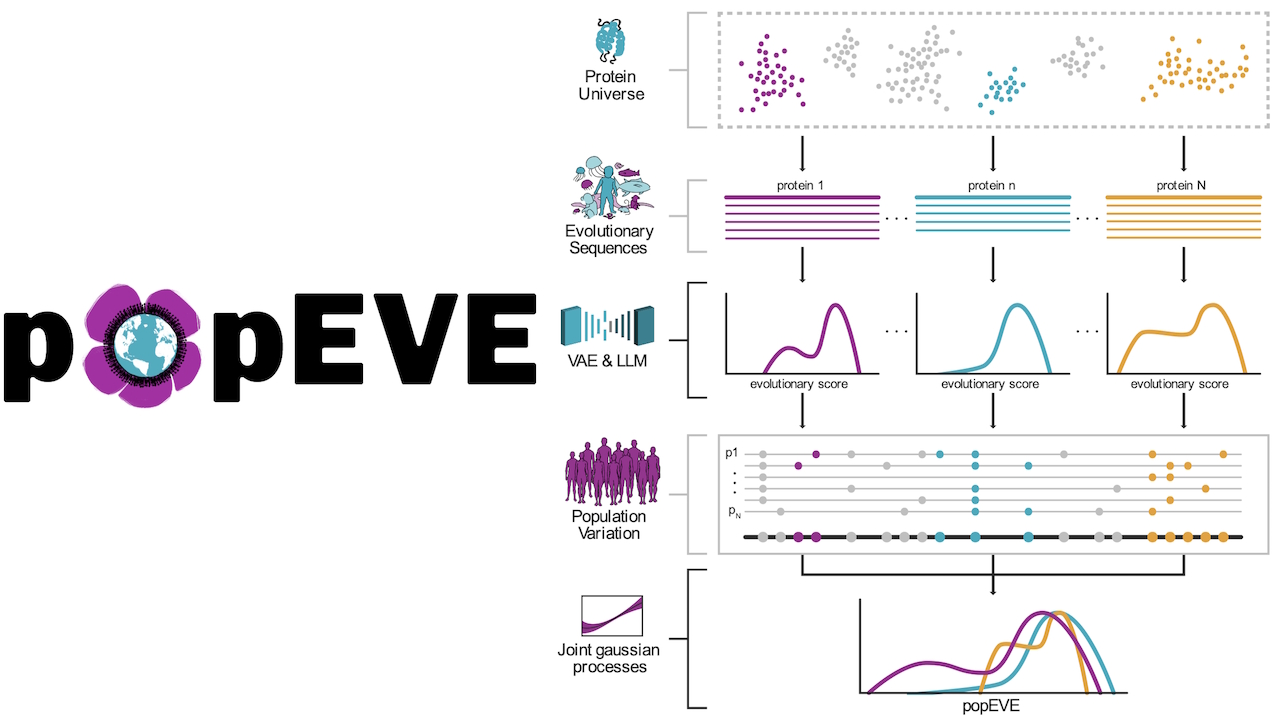
popEVE rozwiązuje ten problem, łącząc głębokie dane ewolucyjne (przy użyciu EVE i modelu języka ESM-1v) z ograniczeniami populacji ludzkiej. Aby określić naturalnie tolerowane warianty, zespół wykorzystał dane z brytyjskiego Biobanku (UKBB) i gnomAD v2.
Do kalibracji wyników ewolucyjnych względem obserwowanej zmienności ludzkiej zastosowano ukryty proces Gaussa, tworząc ujednolicony wynik „szkodliwości”. Dzięki tej korekcie możliwy staje się przełom kliniczny: analiza „pojedyncza”, w której można ustalić priorytety wariantów przyczynowych na podstawie wyłącznie egzomu dziecka.
Tradycyjne metody zazwyczaj wymagają sekwencjonowania „trio” (rodzice + dziecko) w celu zidentyfikowania mutacji de novo, co jest procesem często zbyt kosztownym lub logistycznie niemożliwym.
Mafalda Dias, badaczka w Centrum Regulacji Genomu, podkreślił praktyczne implikacje tej możliwości. „Kliniki nie zawsze mają dostęp do rodzicielskiego DNA i wielu pacjentów przychodzi samotnie. popEVE może pomóc tym lekarzom zidentyfikować mutacje powodujące chorobę.”
Wyzwanie AlphaMissense
AlfaMissense firmy Google DeepMind, wydana we wrześniu 2023 r., wcześniej ustanowiła nowy standard, kategoryzując 89% wszystkich możliwych wariantów zmiany sensu. Zespół z Harvardu argumentuje jednak, że chociaż AlphaMissense pozwala dokładnie określić stabilność białka, brakuje mu kalibracji klinicznej niezbędnej do postawienia diagnozy.
Analiza statystyczna pokazuje, że AlphaMissense przewiduje średnio pięć wariantów „patogennych” na przeciętnego człowieka, podczas gdy popEVE przewiduje mniej niż jeden. Taka rozbieżność jest niezbędna w warunkach klinicznych, gdzie przesadne przewidywanie może prowadzić do błędnej diagnozy i niepotrzebnego niepokoju.
W artykule PrpopEVE dodatkowo zauważono:
„popEVE identyfikuje 442 geny w kohorcie zaburzeń rozwojowych, w tym dowody na 123 nowych kandydatów, z których wiele nie wymaga wzbogacania całej kohorty.”
„Na koniec pokazujemy, że te ustalenia można odtworzyć na podstawie analiza samych egzomów pacjentów, co pokazuje, że popEVE zapewnia nową drogę analizy genetycznej w sytuacjach, w których zawodzą tradycyjne metody.”
Pomimo wzrostu wydajności, popEVE pozostaje narzędziem badawczym i nie uzyskał jeszcze zezwolenia FDA na stosowanie jako samodzielnego urządzenia diagnostycznego. Marks Lab udostępnia model za pośrednictwem otwartego portalu popEVE i repozytorium popEVE, co kontrastuje z często zastrzeżonym charakterem komercyjnych narzędzi AI w zakresie zdrowia.
Przyszłe zastosowania wykraczają poza diagnostykę i odkrywanie leków, jak może to zrobić model wskazać konkretne mechanizmy chorobotwórcze w strukturach białkowych.
Rose Orenbuch, pracownik naukowy w Marks Lab, wyraziła optymizm co do integracji narzędzia z procesami klinicznymi. „Mam wrażenie, że jesteśmy o krok bliżej tego, by popEVE było przydatne w codziennym procesie szybszego diagnozowania chorób genetycznych.”
