計算生物学における Google DeepMind の優位性に対抗して、ハーバード大学医学部の研究者は、希少な遺伝性疾患を特異性を高めて診断するように設計された新しい人工知能モデル、popEVE を発表しました。
本日 Nature Genetics に掲載されたこのツールは、人口データを統合して、AlphaMissense などの既存のモデルの永続的な欠陥である誤検知予測を大幅に削減します。
全体にわたるバリアントの重大度を調整することにより、プロテオーム、popEVE は、発達障害の 123 個の新規候補遺伝子の同定に成功し、広範な検査にもかかわらず未解決のままである患者に診断上の画期的な手段を提供しました。
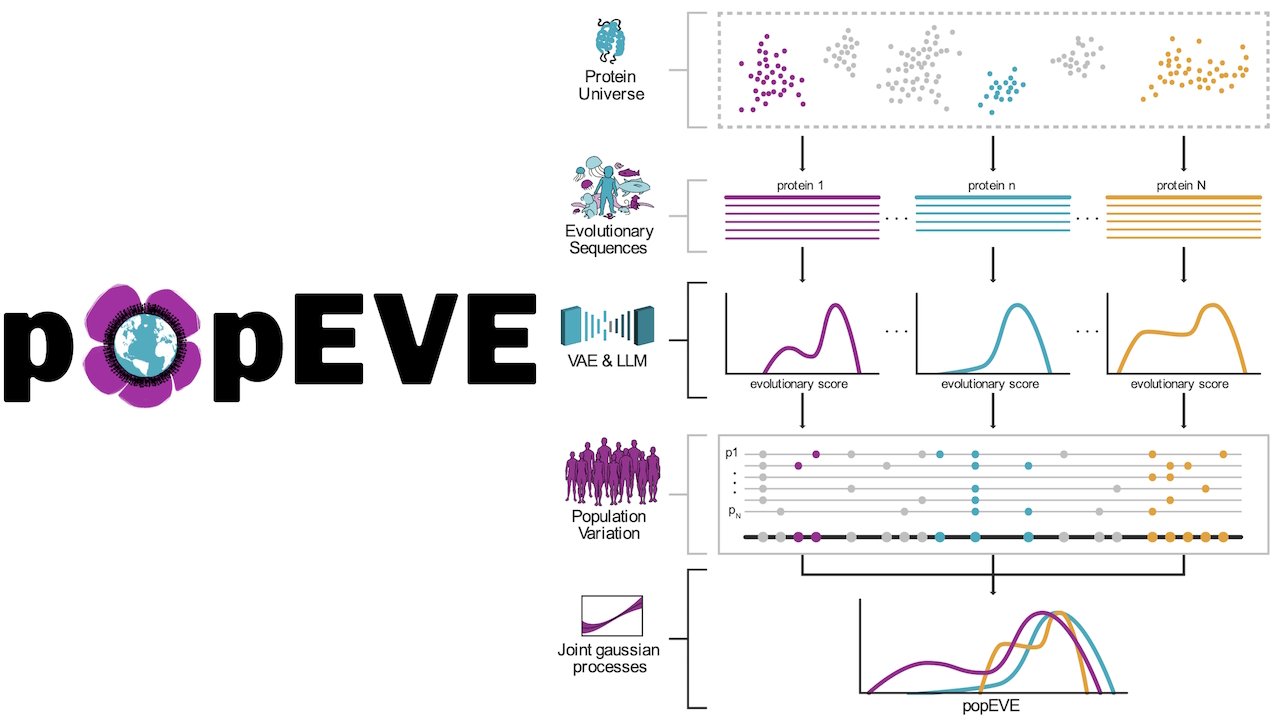
popEVE は偽陽性問題の解決を目指しています
臨床現場におけるゲノム配列決定の急速な拡大にもかかわらず、まれな遺伝性疾患の診断率は依然として低いままであり、一部のコホートでは、発端者のわずか 25% しか最終的な遺伝子診断を受けていないことがわかります。
臨床医は、ヒトの健康への影響が不明な遺伝子変異である膨大な数の「意義不明変異体」(VUS) に頻繁に直面しています。
この曖昧さが、診断のボトルネックを生み出しています。患者の状態の原因となる特定の変異を特定することは、時間がかかり、多くの場合無駄な作業となります。現在の解釈では、小児期に発症する重篤な障害を引き起こすバリアントと、小児医療にとって重要な区別である、小児期に発症する軽度の影響を伴うバリアントを区別できないことがよくあります。
研究論文によると、popEVE は、より厳格なしきい値を適用することで、この精度のギャップに対処しています。病原性。テストでは、このモデルは一般集団内での偽陽性予測が劇的に減少することを実証し、わずか 11% の人に重篤なバリアントの保因者としてフラグを立てました。
このレベルの特異性は、既存の最先端ツールと比較して顕著な改善です。たとえば、Google DeepMind の AlphaMissense は、一般人口の約 44% が、同等の再現閾値で同様に重篤な変異を保有していると分類しています。このノイズをフィルタリングすることで、popEVE を使用すると、臨床医は最も因果関係がある可能性が高い変異に焦点を当てることができます。
このモデルの有効性は、発達障害解読 (DDD) 研究、GeneDx、およびラドバウド大学医療センターから得た重度発達障害 (SDD) 患者 31,058 人のメタコホートで厳密に検証されました。
この広範なデータセット内で、 PopEVE の高信頼性重症度閾値 (-5.056 に設定) により、病原性バリアントが 15 倍濃縮されていることが明らかになりました。これは、PrimateAI-3D などの他の主要な手法よりも 5 倍高い値です。この統計的検出力により、このモデルは、これまで標準的な検査プロトコルでは説明できなかった症例の約 3 分の 1 について診断を下すことに成功しました。
おそらく、医療遺伝学の分野にとって最も重要なのは、まったく新しい疾患の関連性を明らかにするモデルの能力です。この分析により、発達障害に関連する 123 個の新規候補遺伝子が特定され、そのうち 119 個は単一変異体レベルで特定可能でした。
ヒト疾患遺伝学のプロテオーム全体モデル
(出典: Nature – CC BY-NC-ND 4.0)
注目すべきことに、これらの遺伝子のうち 31 個はミスセンス変異のみを使用して回収されました。ミスセンス変異は、通常、診断とみなされる機能喪失 (LoF) データの裏付けを必要とする変異のカテゴリーです。この機能は、popEVE が従来の濃縮ベースの方法では見逃していた病原性シグナルを検出できることを示唆しています。
これらの発見の検証はすでに臨床結果をもたらしています。研究の開始以来、123 個の新規候補遺伝子のうち 25 個が他の研究室によって独立して確認され、発達障害遺伝子表現型 (DDG2P) データベースに正式に追加されました。
さらに、de novo ミスセンス変異 (DNM) に適用した場合、このモデルは、健常対照ではわずか 0.5% であったのに対し、症例では 7% の変異に重度のフラグを立て、高度なフラグメントを示しました。
ハーバード大学医学部のシステム生物学教授であるデボラ マークス氏は、このツールがこれらの統計的利益を具体的な臨床結果に変換するように設計されていることを強調しました。 「私たちの目標は、病気の重症度によって変異をランク付けするモデルを開発し、人のゲノムについて臨床的に意味のある優先順位の高いビューを提供することです。」
プロテオームのキャリブレーション
EVE や AlphaMissense などのこれまでの最先端のモデルは、単一遺伝子内の変異のランク付けには優れていますが、異なる遺伝子間の重症度を比較するのは困難です。その結果、タンパク質の機能を破壊するものの、人間の状況では必ずしも重篤な疾患を引き起こすわけではないバリアントに対して高いスコアが表示されることがよくあります。
popEVE は、深い進化データ (EVE と ESM-1v 言語モデルを使用) を人間の人口制約と組み合わせることで、これを解決します。自然に許容される変異を決定するために、チームは英国バイオバンク (UKBB) と gnomAD v2 からのデータを利用しました。
潜在ガウス プロセスを使用して、この観察された人間の変異に対して進化スコアを調整し、統一された「有害性」スコアを作成します。この調整により、子供のエクソームのみを使用して原因変異の優先順位を付けることができる「シングルトン」解析という大きな臨床的進歩が可能になります。
従来の方法では通常、de novo 突然変異を特定するために「トリオ」シーケンス (親 + 子) が必要ですが、このプロセスは法外に高価であるかロジスティック的に不可能であることがよくあります。
ゲノム制御センターの研究者であるマファルダ ディアス氏は、この機能の実際的な意味を強調しました。 「クリニックは常に親の DNA にアクセスできるわけではなく、多くの患者は一人で来院します。popEVE は、こうした医師が病気の原因となる変異を特定するのに役立ちます。」
挑戦的な AlphaMissense
2023 年 9 月にリリースされた Google DeepMind の AlphaMissense は、以前、考えられるすべてのミスセンス変異体の 89% を分類することで新しい基準を設定しました。しかし、ハーバード大学のチームは、AlphaMissense はタンパク質の安定性については正確であるものの、診断に必要な臨床校正が欠けていると主張しています。
統計分析によると、AlphaMissense は平均的な人あたり平均 5 つの「病原性」バリアントを予測するのに対し、popEVE は 1 つ未満を予測します。このような不一致は、過剰予測が誤診や不必要な不安につながる可能性がある臨床現場では極めて重要です。
PrpopEVE の論文ではさらに次のように述べられています。
「popEVE は、123 の新規候補の証拠を含む、発達障害コホート内の 442 個の遺伝子を特定し、その多くはコホート全体の濃縮を必要としない。」
「最後に、これらの発見は再現可能であることを示す」患者のエクソームのみの分析から、従来の方法が失敗する状況において、popEVE が遺伝子分析に新たな手段を提供することが実証されました。」
性能は向上しているにもかかわらず、popEVE は依然として研究ツールであり、スタンドアロンの診断デバイスとして使用するための FDA の認可をまだ受けていません。 Marks Lab は、このモデルをオープンな popEVE ポータル および popEVE リポジトリ経由で利用できるようにしています。これは、商用 AI 健康ツールの専有的な性質が多いのとは対照的です。
このモデルは特定の特定のポイントを特定できるため、将来のアプリケーションは診断を超えて創薬まで拡張されます。
Marks Lab の研究員である Rose Orenbuch 氏は、このツールの臨床ワークフローへの統合について楽観的な見方を表明しました。 「遺伝病をより迅速に診断するという日々のパイプラインにおいて、popEVE が役立つことにまた一歩近づいたように感じます。」