Sfidando il predominio di Google DeepMind nella biologia computazionale, i ricercatori della Harvard Medical School hanno svelato popEVE, un nuovo modello di intelligenza artificiale progettato per diagnosticare malattie genetiche rare con maggiore specificità.
Pubblicato oggi su Nature Genetics, lo strumento integra i dati della popolazione umana per ridurre drasticamente le previsioni false positive, un difetto persistente nei modelli esistenti come AlphaMissense.
Calibrando la gravità delle varianti nell’intero proteoma, popEVE ha identificato con successo 123 nuovi geni candidati per i disturbi dello sviluppo, offrendo una svolta diagnostica per i pazienti che sono rimasti irrisolti nonostante test approfonditi.
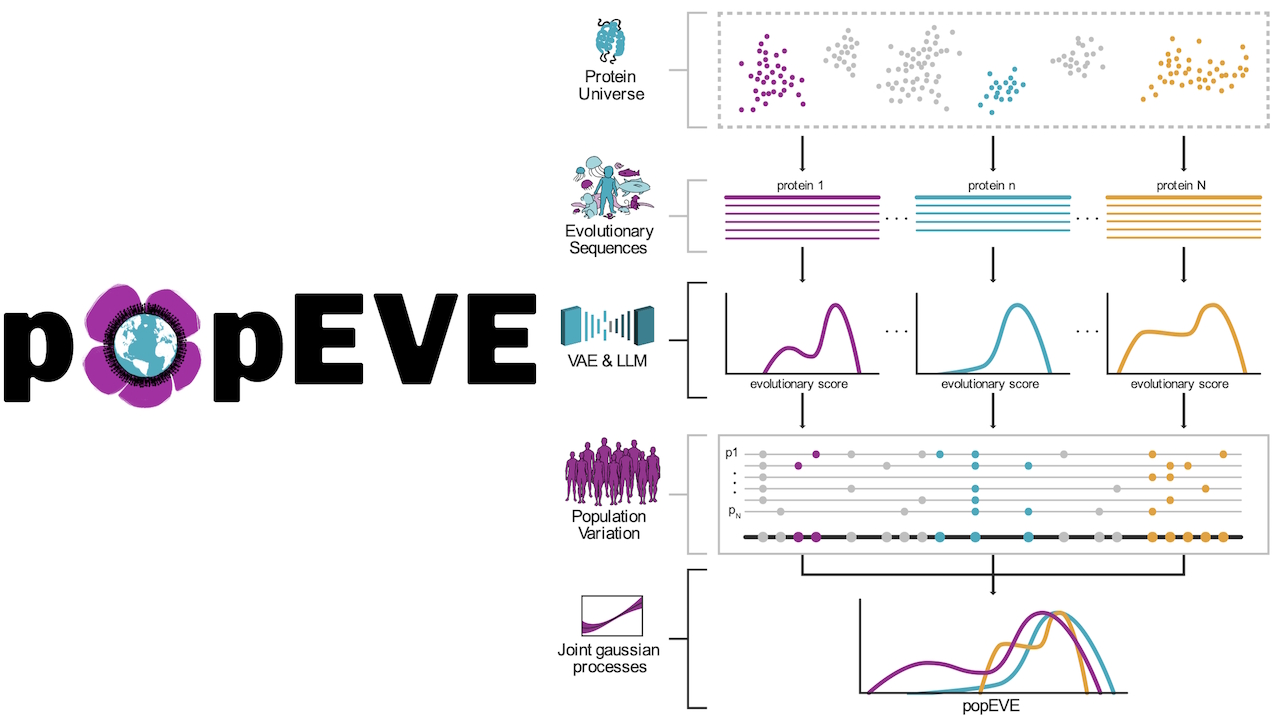
popEVE mira a risolvere il problema dei falsi positivi
Nonostante la rapida espansione del sequenziamento genomico in contesti clinici, il risultato diagnostico per le malattie genetiche rare rimane ostinatamente basso, con alcune coorti che vedono che solo il 25% dei probandi riceve una diagnosi genetica definitiva.
I medici si trovano spesso di fronte a una vasta gamma di”Varianti di significato incerto”(VUS), alterazioni genetiche il cui impatto sulla salute umana è sconosciuto.
Questa ambiguità crea un collo di bottiglia diagnostico, dove identificare la variante specifica responsabile della condizione di un paziente diventa uno sforzo lungo e spesso infruttuoso. L’interpretazione attuale spesso non riesce a distinguere tra le varianti che causano gravi disturbi a esordio infantile e quelle con effetti modesti che si manifestano solo più tardi nella vita, una distinzione fondamentale per l’assistenza pediatrica.
Secondo il documento di ricerca, popEVE affronta questo divario di precisione imponendo una soglia più rigorosa per la patogenicità. Durante i test, il modello ha dimostrato una drastica riduzione delle previsioni false positive nella popolazione generale, segnalando solo l’11% degli individui come portatori di varianti gravi.
Questo livello di specificità rappresenta un netto miglioramento rispetto agli strumenti all’avanguardia esistenti; ad esempio, AlphaMissense di Google DeepMind classifica circa il 44% della popolazione generale come portatrice di varianti altrettanto gravi a soglie di richiamo comparabili. Filtrando questo rumore, popEVE consente ai medici di concentrarsi sulle varianti che hanno più probabilità di essere causali.
L’efficacia del modello è stata rigorosamente convalidata su una metacoorte di 31.058 pazienti con gravi disturbi dello sviluppo (SDD), provenienti dallo studio Deciphering Developmental Disorders (DDD), GeneDx e Radboud University Medical Center.
All’interno di questo ampio set di dati, popEVE la soglia di gravità ad alta confidenza (fissata a-5.056) ha rivelato un arricchimento di 15 volte di varianti patogene, cinque volte superiore rispetto ad altri metodi leader come PrimateAI-3D. Questa potenza statistica ha consentito al modello di fornire con successo una diagnosi per circa un terzo dei casi che in precedenza non potevano essere spiegati con protocolli di test standard.
Forse la cosa più significativa per il campo della genetica medica è la capacità del modello di scoprire associazioni di malattie completamente nuove. L’analisi ha identificato 123 nuovi geni candidati collegati a disturbi dello sviluppo, 119 dei quali erano identificabili a livello di singola variante.
Modello a livello di proteoma per la genetica delle malattie umane
(Fonte: Nature – CC BY-NC-ND 4.0)
In particolare, 31 di questi geni sono stati recuperati utilizzando solo varianti missenso, una categoria di mutazione che in genere richiede dati corroboranti sulla perdita di funzione (LoF) per essere considerata diagnostica. Questa capacità suggerisce che popEVE può rilevare segnali patogeni che i metodi tradizionali basati sull’arricchimento non rilevano.
La convalida di questi risultati sta già producendo risultati clinici. Dall’inizio dello studio, 25 dei 123 nuovi geni candidati sono stati confermati in modo indipendente da altri laboratori e formalmente aggiunti al database Developmental Disorder Gene to Phenotype (DDG2P).
Inoltre, quando applicato alle mutazioni missenso de novo (DNM), il modello ha contrassegnato il 7% delle varianti nei casi gravi, rispetto a solo lo 0,5% nei controlli sani, dimostrando un alto grado di separazione tra patogeni e benigni. variazioni.
Debora Marks, professoressa di biologia dei sistemi presso la Harvard Medical School, ha sottolineato che lo strumento è progettato per tradurre questi miglioramenti statistici in risultati clinici tangibili.”Il nostro obiettivo era sviluppare un modello che classifichi le varianti in base alla gravità della malattia, fornendo una visione prioritaria e clinicamente significativa del genoma di una persona.”
Calibrazione del proteoma
Precedenti modelli all’avanguardia, tra cui EVE e AlphaMissense, eccellono nel classificare le varianti all’interno di un singolo gene ma hanno difficoltà a confrontare la gravità tra geni diversi. Di conseguenza, punteggi elevati appaiono spesso per varianti che interrompono la funzione proteica ma non necessariamente causano malattie gravi in un contesto umano.
popEVE risolve questo problema combinando dati evolutivi approfonditi (utilizzando EVE e il modello linguistico ESM-1v) con i vincoli della popolazione umana. Per determinare le varianti naturalmente tollerate, il team ha utilizzato i dati della UK Biobank (UKBB) e di gnomAD v2.
Un processo gaussiano latente viene impiegato per calibrare i punteggi evolutivi rispetto a questa variazione umana osservata, creando un punteggio unificato di”dannosità”. Attraverso questo aggiustamento, diventa possibile un importante passo avanti clinico: l’analisi”singleton”, in cui è possibile dare priorità alle varianti causali utilizzando solo l’esoma del bambino.
I metodi tradizionali richiedono in genere il sequenziamento”trio”(genitori + figlio) per identificare le mutazioni de novo, un processo che spesso è proibitivamente costoso o logisticamente impossibile.
Mafalda Dias, ricercatrice presso il Center for Genomic Adjustment, ha evidenziato le implicazioni pratiche di questa capacità.”Le cliniche non hanno sempre accesso al DNA dei genitori e molti pazienti vengono da soli. popEVE può aiutare questi medici a identificare le mutazioni che causano malattie.”
La sfida di AlphaMissense
AlphaMissense di Google DeepMind, pubblicato nel settembre 2023, aveva precedentemente stabilito un nuovo standard classificando l’89% di tutte le possibili varianti missenso. Tuttavia, il team di Harvard sostiene che, sebbene AlphaMissense sia accurato per la stabilità delle proteine, manca della calibrazione clinica necessaria per la diagnosi.
L’analisi statistica mostra che AlphaMissense predice una media di cinque varianti”patogene”per persona media, mentre popEVE ne prevede meno di una. Tale discrepanza è vitale per i contesti clinici, dove una previsione eccessiva può portare a diagnosi errate e ansia non necessaria.
Il documento PrpopEVE rileva inoltre:
“popEVE identifica 442 geni in una coorte di disturbi dello sviluppo, inclusa l’evidenza di 123 nuovi candidati, molti senza la necessità di un arricchimento a livello di coorte.”
“Infine, mostriamo che questi risultati possono essere riprodotti dall’analisi di il paziente esegue l’esame da solo, dimostrando che popEVE fornisce una nuova strada per l’analisi genetica in situazioni in cui i metodi tradizionali falliscono.”
Nonostante i miglioramenti in termini di prestazioni, popEVE rimane uno strumento di ricerca e non ha ancora ricevuto l’autorizzazione della FDA per l’uso come dispositivo diagnostico autonomo. Marks Lab sta rendendo disponibile il modello tramite un portale popEVE aperto e un repository popEVE, in contrasto con la natura spesso proprietaria degli strumenti sanitari commerciali basati sull’intelligenza artificiale.
Le applicazioni future vanno oltre la diagnosi fino alla scoperta di farmaci, poiché il modello può individuare specifici meccanismi patogeni all’interno delle proteine strutture.
Rose Orenbuch, ricercatrice presso il Marks Lab, ha espresso ottimismo riguardo all’integrazione dello strumento nei flussi di lavoro clinici.”Sento che siamo un passo avanti verso il fatto che popEVE possa essere utile nel tentativo quotidiano di diagnosticare le malattie genetiche più velocemente.”