Contestant la domination de Google DeepMind en biologie computationnelle, des chercheurs de la Harvard Medical School ont dévoilé popEVE, un nouveau modèle d’intelligence artificielle conçu pour diagnostiquer les maladies génétiques rares avec une spécificité améliorée.
Publié aujourd’hui dans Nature Genetics, l’outil intègre des données sur la population humaine pour réduire considérablement les prédictions faussement positives, un défaut persistant dans les modèles existants comme AlphaMissense.
En calibrant la gravité des variantes sur l’ensemble du protéome, popEVE a identifié avec succès 123 nouveaux gènes candidats pour les troubles du développement, offrant une avancée diagnostique pour les patients qui sont restés non résolus malgré des tests approfondis.
popEVE vise à résoudre le problème des faux positifs
Malgré l’expansion rapide du séquençage génomique en milieu clinique, le rendement du diagnostic pour les maladies génétiques rares reste obstinément faible, certaines cohortes voyant seulement 25 % des proposants recevoir un diagnostic génétique définitif.
Les cliniciens sont fréquemment confrontés à un vaste éventail de « variantes de signification incertaine » (VUS), des altérations génétiques dont l’impact sur la santé humaine est inconnu.
Cette ambiguïté crée un goulot d’étranglement diagnostique, où l’identification de la variante spécifique responsable de l’état d’un patient devient une entreprise longue et souvent infructueuse. L’interprétation actuelle ne parvient souvent pas à faire la distinction entre les variantes qui provoquent des troubles graves apparaissant dans l’enfance et celles ayant des effets modestes qui ne se manifestent que plus tard dans la vie, une distinction essentielle pour les soins pédiatriques.
Selon le document de recherche, popEVE comble cet écart de précision en imposant un seuil de pathogénicité plus strict. Lors des tests, le modèle a démontré une réduction spectaculaire des prédictions faussement positives au sein de la population générale, signalant seulement 11 % des individus comme porteurs de variantes graves.
Ce niveau de spécificité constitue une nette amélioration par rapport aux outils de pointe existants ; par exemple, AlphaMissense de Google DeepMind classe environ 44 % de la population générale comme étant porteuse de variantes tout aussi graves à des seuils de rappel comparables. En filtrant ce bruit, popEVE permet aux cliniciens de se concentrer sur les variantes les plus susceptibles d’être causales.
L’efficacité du modèle a été rigoureusement validée sur une métacohorte de 31 058 patients atteints de troubles graves du développement (SDD), provenant de l’étude Deciphering Developmental Disorders (DDD), GeneDx et Radboud University Medical Center.
Au sein de ce vaste ensemble de données, la gravité de haut niveau de confiance de popEVE Le seuil (fixé à-5,056) a révélé un enrichissement de 15 fois en variantes pathogènes – cinq fois plus élevé que d’autres méthodes de pointe telles que PrimateAI-3D. Cette puissance statistique a permis au modèle de fournir avec succès un diagnostic pour environ un tiers des cas qui avaient auparavant défié toute explication selon les protocoles de test standard.
Le plus important peut-être pour le domaine de la génétique médicale est la capacité du modèle à découvrir des associations de maladies entièrement nouvelles. L’analyse a identifié 123 nouveaux gènes candidats liés à des troubles du développement, dont 119 étaient identifiables au niveau d’une seule variante.
Modèle à l’échelle du protéome pour la génétique des maladies humaines
(Source : Nature – CC BY-NC-ND 4.0)
Notamment, 31 de ces gènes ont été récupérés en utilisant uniquement des variantes faux-sens-une catégorie de mutation qui nécessite généralement des données corroborantes de perte de fonction (LoF) pour être considérée comme un diagnostic. Cette capacité suggère que popEVE peut détecter les signaux pathogènes qui manquent aux méthodes traditionnelles basées sur l’enrichissement.
La validation de ces résultats donne déjà des résultats cliniques. Depuis le début de l’étude, 25 des 123 nouveaux gènes candidats ont été confirmés de manière indépendante par d’autres laboratoires et officiellement ajoutés à la base de données DDG2P (Developmental Disorder Gene to Phenotype).
De plus, lorsqu’il est appliqué à des mutations faux-sens (DNM) de novo, le modèle a signalé 7 % des variants dans les cas comme étant graves, contre seulement 0,5 % chez les témoins sains, démontrant un degré élevé de séparation entre pathogènes et variations bénignes.
Debora Marks, professeur de biologie des systèmes à la Harvard Medical School, a souligné que l’outil est conçu pour traduire ces gains statistiques en résultats cliniques tangibles.”Notre objectif était de développer un modèle qui classe les variantes par gravité de la maladie, fournissant ainsi une vue prioritaire et cliniquement significative du génome d’une personne.”
Calibrage du protéome
Les modèles de pointe précédents, notamment EVE et AlphaMissense, excellent dans le classement des variantes au sein d’un seul gène, mais ont du mal à comparer la gravité entre différents gènes. Par conséquent, des scores élevés apparaissent souvent pour les variantes qui perturbent la fonction des protéines mais ne provoquent pas nécessairement de maladies graves dans un contexte humain.
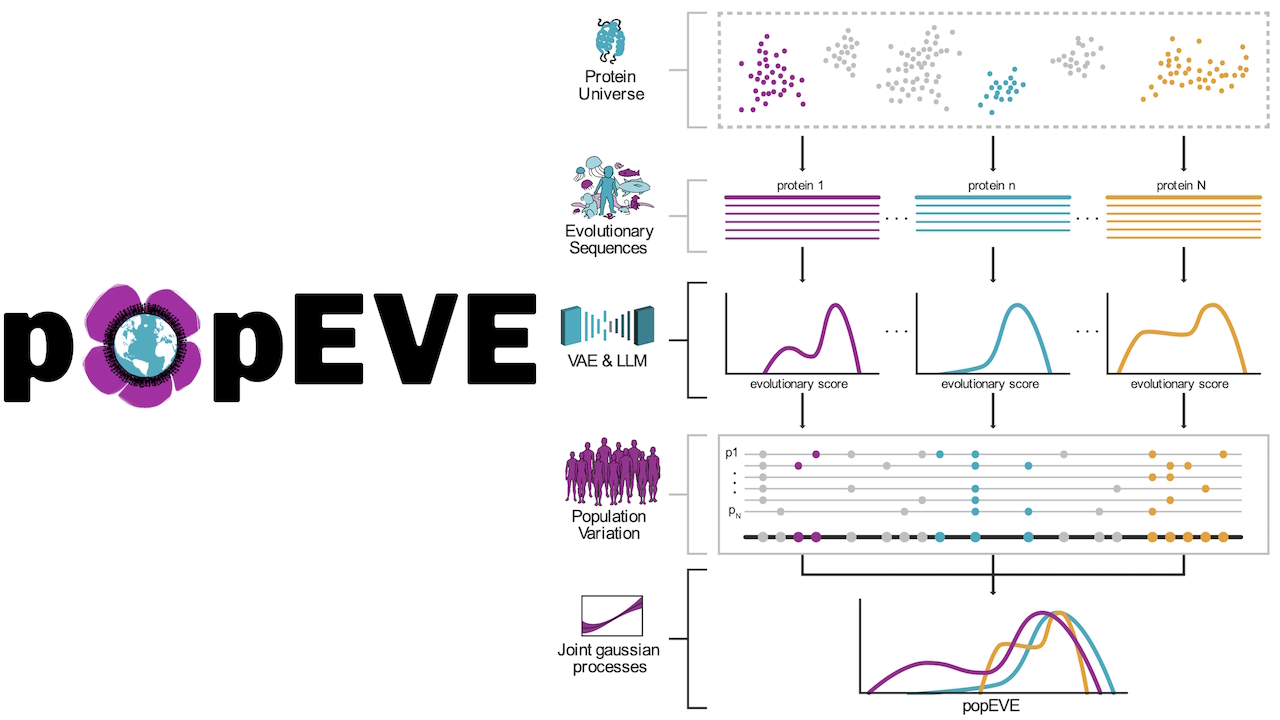
popEVE résout ce problème en combinant des données évolutives approfondies (en utilisant EVE et le modèle de langage ESM-1v) avec les contraintes de la population humaine. Pour déterminer les variantes naturellement tolérées, l’équipe a utilisé les données de la UK Biobank (UKBB) et de gnomAD v2.
Un processus gaussien latent est utilisé pour calibrer les scores évolutifs par rapport à cette variation humaine observée, créant ainsi un score unifié de « caractère délétère ». Grâce à cet ajustement, une avancée clinique majeure devient possible: l’analyse « singleton », où les variantes causales peuvent être priorisées en utilisant uniquement l’exome de l’enfant.
Les méthodes traditionnelles nécessitent généralement un séquençage en « trio » (parents + enfant) pour identifier les mutations de novo, un processus qui est souvent d’un coût prohibitif ou impossible sur le plan logistique.
Mafalda Dias, chercheuse au Centre de régulation génomique, a souligné les implications pratiques de cette capacité.”Les cliniques n’ont pas toujours accès à l’ADN parental et de nombreux patients viennent seuls. popEVE peut aider ces médecins à identifier les mutations causant des maladies.”
Défier AlphaMissense
AlphaMissense de Google DeepMind, publié en septembre 2023, avait déjà établi une nouvelle norme en catégorisant 89 % de toutes les variantes faux-sens possibles. Cependant, l’équipe de Harvard affirme que même si AlphaMissense est précis pour la stabilité des protéines, il lui manque l’étalonnage clinique nécessaire au diagnostic.
L’analyse statistique montre qu’AlphaMissense prédit une moyenne de cinq variantes « pathogènes » par personne moyenne, alors que popEVE en prédit moins d’une. Un tel écart est vital en milieu clinique, où une surprédiction peut conduire à des diagnostics erronés et à une anxiété inutile.
L’article PrpopEVE note en outre :
“popEVE identifie 442 gènes dans une cohorte de troubles du développement, y compris la preuve de 123 nouveaux candidats, dont beaucoup sans avoir besoin d’un enrichissement à l’échelle de la cohorte.”
“Enfin, nous montrons que ces résultats peuvent être reproduits à partir de l’analyse des exomes des patients. à lui seul, démontrant que popEVE offre une nouvelle voie pour l’analyse génétique dans les situations où les méthodes traditionnelles échouent.”
Malgré les gains de performances, popEVE reste un outil de recherche et n’a pas encore reçu l’autorisation de la FDA pour une utilisation comme dispositif de diagnostic autonome. Marks Lab rend le modèle disponible via un portail popEVE ouvert et un dépôt popEVE, ce qui contraste avec la nature souvent exclusive des outils commerciaux de santé liés à l’IA.
Les applications futures s’étendent au-delà du diagnostic jusqu’à la découverte de médicaments, car le modèle peut identifier des agents pathogènes spécifiques. mécanismes au sein des structures protéiques.
Rose Orenbuch, chercheuse au Marks Lab, a exprimé son optimisme quant à l’intégration de l’outil dans les flux de travail cliniques.”J’ai l’impression que nous sommes sur le point de permettre à popEVE d’être utile au quotidien pour essayer de diagnostiquer plus rapidement les maladies génétiques.”


