Desafiando el dominio de Google DeepMind en biología computacional, investigadores de la Facultad de Medicina de Harvard han presentado popEVE, un nuevo modelo de inteligencia artificial diseñado para diagnosticar enfermedades genéticas raras con especificidad mejorada.
Publicada hoy en Nature Genetics, la herramienta integra datos de población humana para reducir drásticamente las predicciones falsas positivas, un defecto persistente en modelos existentes como AlphaMissense.
Al calibrar la gravedad de las variantes en todo el proteoma, popEVE tuvo éxito. identificó 123 nuevos genes candidatos para trastornos del desarrollo, lo que ofrece un avance diagnóstico para pacientes que siguen sin resolverse a pesar de pruebas exhaustivas.
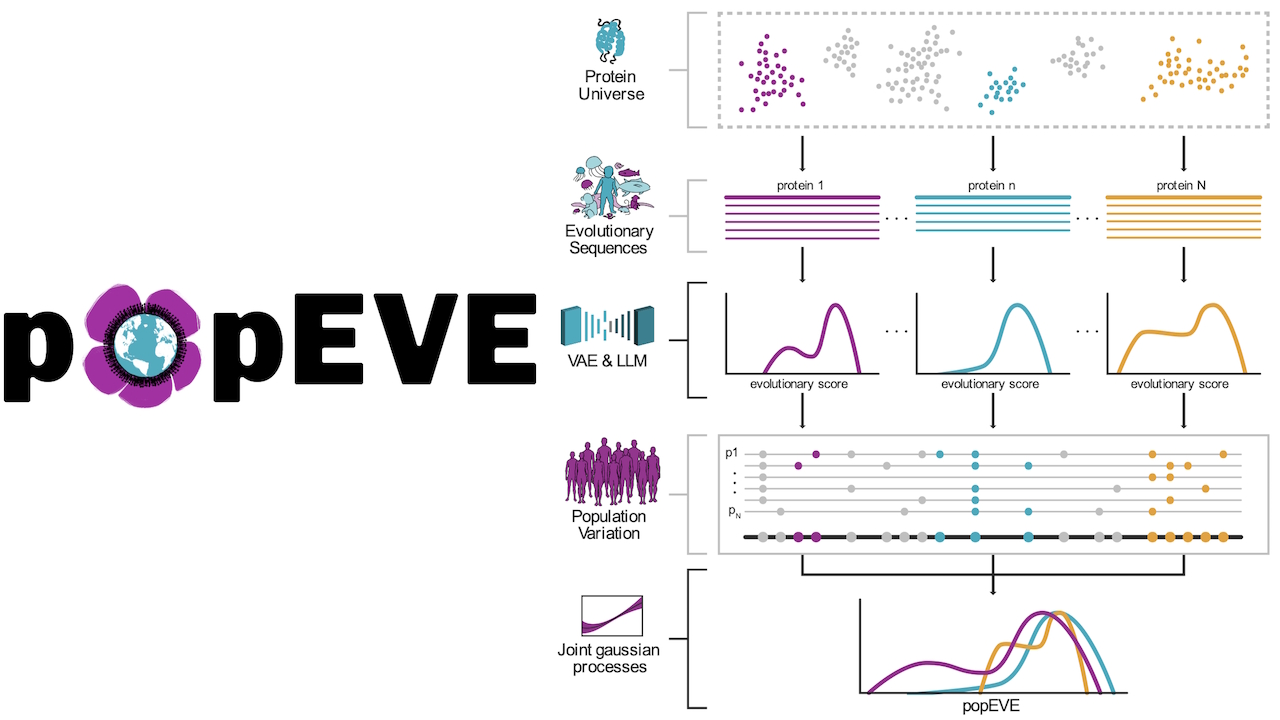
popEVE apunta a resolver el problema de los falsos positivos
A pesar de la rápida expansión de la secuenciación genómica en entornos clínicos, el rendimiento diagnóstico El número de trastornos genéticos raros sigue siendo obstinadamente bajo, y en algunas cohortes solo el 25% de los probandos reciben un diagnóstico genético definitivo.
Los médicos se enfrentan con frecuencia a una amplia gama de”variantes de significado incierto”(VUS), alteraciones genéticas cuyo impacto en la salud humana se desconoce.
Esta ambigüedad crea un cuello de botella en el diagnóstico, donde identificar la variante específica responsable de la condición de un paciente se convierte en un esfuerzo que requiere mucho tiempo y, a menudo, infructuoso. La interpretación actual a menudo no distingue entre las variantes que causan trastornos graves que comienzan en la niñez y aquellas con efectos modestos que se manifiestan solo más adelante en la vida, una distinción crítica para la atención pediátrica.
Según el artículo de investigación, popEVE aborda esta brecha de precisión imponiendo un umbral más estricto para patogenicidad. En las pruebas, el modelo demostró una reducción dramática en las predicciones falsas positivas dentro de la población general, señalando que solo el 11 % de los individuos son portadores de variantes graves.
Este nivel de especificidad es una mejora notable con respecto a las herramientas de última generación existentes; por ejemplo, AlphaMissense de Google DeepMind clasifica a aproximadamente el 44% de la población general como portadora de variantes igualmente graves con umbrales de recuerdo comparables. Al filtrar este ruido, popEVE permite a los médicos centrarse en las variantes con mayor probabilidad de ser causales.
La eficacia del modelo se validó rigurosamente en una metacohorte de 31.058 pacientes con trastornos graves del desarrollo (SDD), provenientes del estudio Deciphering Developmental Disorders (DDD), GeneDx y Radboud University Medical Center.
Dentro de este extenso conjunto de datos, popEVE El umbral de gravedad de alta confianza (establecido en-5,056) reveló un enriquecimiento 15 veces mayor de variantes patógenas, cinco veces mayor que otros métodos líderes como PrimateAI-3D. Este poder estadístico permitió que el modelo proporcionara con éxito un diagnóstico para aproximadamente un tercio de los casos que previamente habían desafiado la explicación según los protocolos de prueba estándar.
Quizás lo más significativo para el campo de la genética médica es la capacidad del modelo para descubrir asociaciones de enfermedades completamente nuevas. El análisis identificó 123 nuevos genes candidatos relacionados con trastornos del desarrollo, 119 de los cuales fueron identificables a nivel de variante única.
Modelo de proteoma completo para la genética de enfermedades humanas
(Fuente: Naturaleza – CC BY-NC-ND 4.0)
En particular, 31 de estos genes se recuperaron utilizando variantes sin sentido únicamente, una categoría de mutación que normalmente requiere corroborar los datos de pérdida de función (LoF) para ser considerada diagnóstica. Esta capacidad sugiere que popEVE puede detectar señales patógenas que los métodos tradicionales basados en el enriquecimiento pasan por alto.
La validación de estos hallazgos ya está arrojando resultados clínicos. Desde el inicio del estudio, 25 de los 123 nuevos genes candidatos han sido confirmados de forma independiente por otros laboratorios y agregados formalmente a la base de datos del gen del trastorno del desarrollo al fenotipo (DDG2P).
Además, cuando se aplicó a mutaciones sin sentido (DNM) de novo, el modelo marcó el 7 % de las variantes en los casos como graves, en comparación con solo el 0,5 % en los controles sanos, lo que demuestra un alto grado de separación entre patógenos y benignos. variaciones.
Debora Marks, profesora de biología de sistemas en la Facultad de Medicina de Harvard, enfatizó que la herramienta está diseñada para traducir estos avances estadísticos en resultados clínicos tangibles.”Nuestro objetivo era desarrollar un modelo que clasificara las variantes según la gravedad de la enfermedad, proporcionando una visión priorizada y clínicamente significativa del genoma de una persona”.
Calibración del proteoma
Los modelos de última generación anteriores, incluidos EVE y AlphaMissense, destacan en clasificar variantes dentro de un solo gen, pero tienen dificultades para comparar la gravedad entre diferentes genes. En consecuencia, a menudo aparecen puntuaciones altas para variantes que alteran la función de las proteínas pero que no necesariamente causan enfermedades graves en un contexto humano.
popEVE resuelve este problema combinando datos evolutivos profundos (utilizando EVE y el modelo de lenguaje ESM-1v) con limitaciones de la población humana. Para determinar las variantes naturalmente toleradas, el equipo utilizó datos del Biobanco del Reino Unido (UKBB) y gnomAD v2.
Se emplea un proceso gaussiano latente para calibrar las puntuaciones evolutivas frente a esta variación humana observada, creando una puntuación unificada de”deletéreo”. A través de este ajuste, se hace posible un gran avance clínico: el análisis”singleton”, donde las variantes causales pueden priorizarse utilizando solo el exoma del niño.
Los métodos tradicionales generalmente requieren una secuenciación”trío”(padres + niño) para identificar mutaciones de novo, un proceso que a menudo es prohibitivamente costoso o logísticamente imposible.
Mafalda Dias, investigadora del Centro de Regulación Genómica, destacó las implicaciones prácticas de esta capacidad.”Las clínicas no siempre tienen acceso al ADN de los padres y muchos pacientes vienen solos. popEVE puede ayudar a estos médicos a identificar mutaciones que causan enfermedades”.
Desafiando AlphaMissense
AlphaMissense de Google DeepMind, lanzado en septiembre de 2023, anteriormente estableció un nuevo estándar al categorizar el 89% de todas las posibles variantes sin sentido. Sin embargo, el equipo de Harvard sostiene que, si bien AlphaMissense es preciso en cuanto a la estabilidad de las proteínas, carece de la calibración clínica necesaria para el diagnóstico.
El análisis estadístico muestra que AlphaMissense predice un promedio de cinco variantes”patógenas”por persona promedio, mientras que popEVE predice menos de una. Esta discrepancia es vital para los entornos clínicos, donde la sobrepredicción puede conducir a diagnósticos erróneos y ansiedad innecesaria.
El artículo PrpopEVE señala además:
“popEVE identifica 442 genes en una cohorte de trastornos del desarrollo, incluida evidencia de 123 candidatos novedosos, muchos de ellos sin necesidad de enriquecimiento en toda la cohorte”.
“Finalmente, demostramos que estos hallazgos se pueden reproducir a partir del análisis del paciente exomas solos, lo que demuestra que popEVE proporciona una nueva vía para el análisis genético en situaciones en las que los métodos tradicionales fallan”.
A pesar de las mejoras en el rendimiento, popEVE sigue siendo una herramienta de investigación y aún no ha recibido la autorización de la FDA para su uso como dispositivo de diagnóstico independiente. Marks Lab está poniendo el modelo a disposición a través de un portal popEVE y un repositorio popEVE, en contraste con la naturaleza a menudo patentada de las herramientas de salud comerciales de IA.
Las aplicaciones futuras se extenderán más allá del diagnóstico hasta el descubrimiento de fármacos, ya que el modelo puede identificar datos específicos mecanismos patogénicos dentro de las estructuras proteicas.
Rose Orenbuch, investigadora del Marks Lab, expresó optimismo sobre la integración de la herramienta en los flujos de trabajo clínicos.”Siento que estamos un paso más cerca de que popEVE sea útil en el proceso diario de tratar de diagnosticar enfermedades genéticas más rápido”.