哈佛醫學院的研究人員推出了 popEVE,這是一種新的人工智能模型,旨在挑戰 Google DeepMind 在計算生物學領域的主導地位。
該工具今天發表在《Nature Genetics》上,它整合了人類群體數據,可大幅減少誤報預測,這是 AlphaMissense 等現有模型中的一個長期缺陷。
通過校準整個蛋白質組的變異嚴重程度,popEVE 成功識別了變異的嚴重程度。 123 個新的發育障礙候選基因,為經過廣泛測試仍未解決的患者提供了診斷突破。
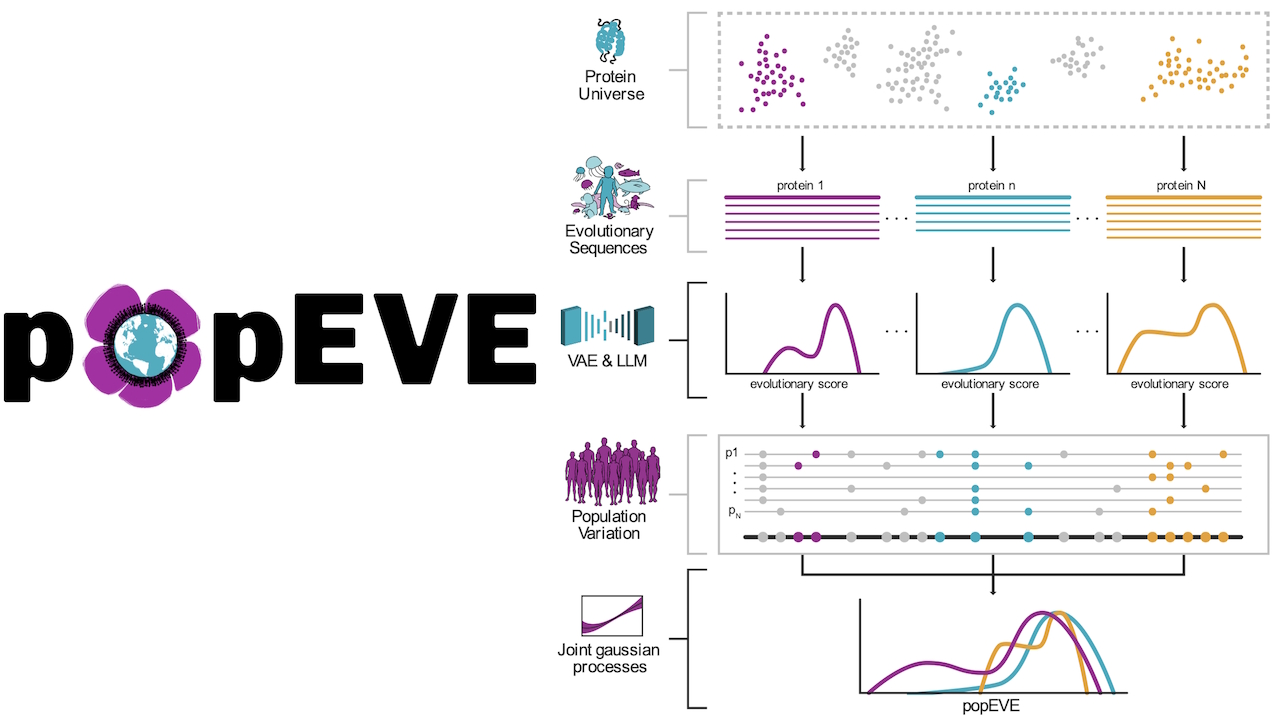
popEVE 旨在解決假陽性問題
儘管基因組測序在臨床環境中迅速擴展,但罕見病的診斷率遺傳性疾病的發病率仍然很低,一些隊列中只有 25% 的先證者獲得了明確的基因診斷。
臨床醫生經常面臨大量“意義不確定的變異”(VUS),這些基因改變對人類健康的影響尚不清楚。
這種模糊性造成了診斷瓶頸,識別導致患者病情的特定變異成為一項耗時且往往徒勞的努力。目前的解釋通常無法區分導致嚴重的兒童期發病疾病的變異和僅在以後的生活中表現出輕微影響的變異,這是兒科護理的一個關鍵區別。
根據研究論文,popEVE 通過強制執行更嚴格的致病性閾值來解決這一精度差距。在測試中,該模型證明了一般人群中假陽性預測的顯著減少,僅將 11% 的個體標記為嚴重變異的攜帶者。
這種特異性水平比現有最先進的工具有了顯著改進;例如,谷歌 DeepMind 的 AlphaMissense 將大約 44% 的普通人群歸類為在可比較的召回閾值下攜帶類似嚴重的變異。通過濾除這些噪音,popEVE 使臨床醫生能夠專注於最有可能造成因果關係的變異。
該模型的功效在 31,058 名患有嚴重發育障礙 (SDD) 患者的元隊列中得到了嚴格驗證,這些患者來自破譯發育障礙 (DDD) 研究、GeneDx 和拉德堡德大學醫學中心。
在這個廣泛的數據集中,popEVE 的高置信度嚴重性閾值(設置為-5.056)顯示致病變異富集了 15 倍——比 PrimateAI-3D 等其他領先方法高出五倍。這種統計能力使該模型能夠成功地為大約三分之一的病例提供診斷,而這些病例以前在標準測試方案下無法解釋。
對於醫學遺傳學領域來說,最重要的也許是該模型能夠發現全新的疾病關聯。該分析確定了 123 個與發育障礙相關的新候選基因,其中 119 個可在單變異水平上識別。
人類疾病遺傳學的蛋白質組範圍模型
(來源:《自然》 – CC BY-NC-ND 4.0)
值得注意的是,其中 31 個基因僅使用錯義變體即可恢復——這種突變通常需要證實功能喪失 (LoF) 數據才能被視為診斷。這種能力表明 popEVE 可以檢測傳統富集方法遺漏的致病信號。
這些發現的驗證已經產生了臨床結果。自研究開始以來,123個新候選基因中的25個已被其他實驗室獨立確認,並正式添加到發育障礙基因表型(DDG2P)數據庫中。
此外,當應用於從頭錯義突變(DNM)時,該模型將病例中7%的變異標記為嚴重,而健康對照中僅為0.5%,這表明致病性變異和良性變異之間存在高度分離。
哈佛醫學院系統生物學教授 Debora Marks強調該工具旨在將這些統計成果轉化為切實的臨床結果。 “我們的目標是開發一種按疾病嚴重程度對變異進行排序的模型,為個人基因組提供優先的、有臨床意義的視圖。”
校準蛋白質組
以前最先進的模型,包括 EVE 和 AlphaMissense,擅長對單個基因內的變異進行排序,但很難比較不同基因的嚴重程度。因此,破壞蛋白質功能但不一定會在人類環境中引起嚴重疾病的變異通常會出現高分。
popEVE 通過將深度進化數據(使用 EVE 和 ESM-1v 語言模型)與人口限制相結合來解決這個問題。為了確定自然耐受的變異,該團隊利用了英國生物銀行 (UKBB) 和 gnomAD v2 的數據。
採用潛在高斯過程來根據觀察到的人類變異來校准進化分數,從而創建統一的“有害性”分數。通過這種調整,重大的臨床突破成為可能:“單例”分析,即僅使用孩子的外顯子組即可對因果變異進行優先排序。
傳統方法通常需要“三重”測序(父母+孩子)來識別從頭突變,這一過程通常成本高昂或在邏輯上是不可能的。
Mafalda Dias,基因組調控中心的研究員,強調了此功能的實際影響。 “診所並不總是能夠獲得父母 DNA,許多患者都是獨自前來。popEVE 可以幫助這些醫生識別緻病突變。”
挑戰 AlphaMissense
Google DeepMind 的 AlphaMissense 於 2023 年 9 月發布,此前通過對所有可能的錯義變異中 89% 進行分類,設定了新標準。然而,哈佛團隊認為,雖然 AlphaMissense 對於蛋白質穩定性來說是準確的,但它缺乏診斷所需的臨床校準。
統計分析顯示 AlphaMissense 預測平均每個人有 5 個“致病”變異,而 popEVE 預測不到 1 個。這種差異對於臨床環境至關重要,過度預測可能會導致誤診和不必要的焦慮。
PrpopEVE 論文進一步指出:
“popEVE 在發育障礙隊列中識別出 442 個基因,包括 123 個新候選基因的證據,其中許多不需要在隊列範圍內進行豐富。”
“最後,我們表明這些發現可以通過對患者的分析來重現單獨的外顯子組,表明 popEVE 在傳統方法失敗的情況下為遺傳分析提供了新途徑。”
儘管性能有所提高,但 popEVE 仍然是一種研究工具,尚未獲得 FDA 批准用作獨立的診斷設備。 Marks Lab 正在通過開放的 popEVE 門戶和 popEVE 存儲庫提供該模型,這與商業人工智能健康工具通常專有的性質形成鮮明對比。
未來的應用範圍將超越診斷,擴展到藥物發現,因為該模型可以查明其中的特定致病機制。
Marks 實驗室研究員 Rose Orenbuch 對該工具融入臨床工作流程表示樂觀。 “我覺得我們距離 popEVE 在嘗試更快地診斷遺傳疾病的日常流程中發揮作用又近了一步。”